



Φαινυλκετονουρία. Η φαινυλκετονουρία είναι μία σχετικά σπάνια κληρονομική ασθένεια και ο οργανισμός δε μπορεί να διασπάσει τη φαινυλαλανίνη
Φαινυλκετονουρία
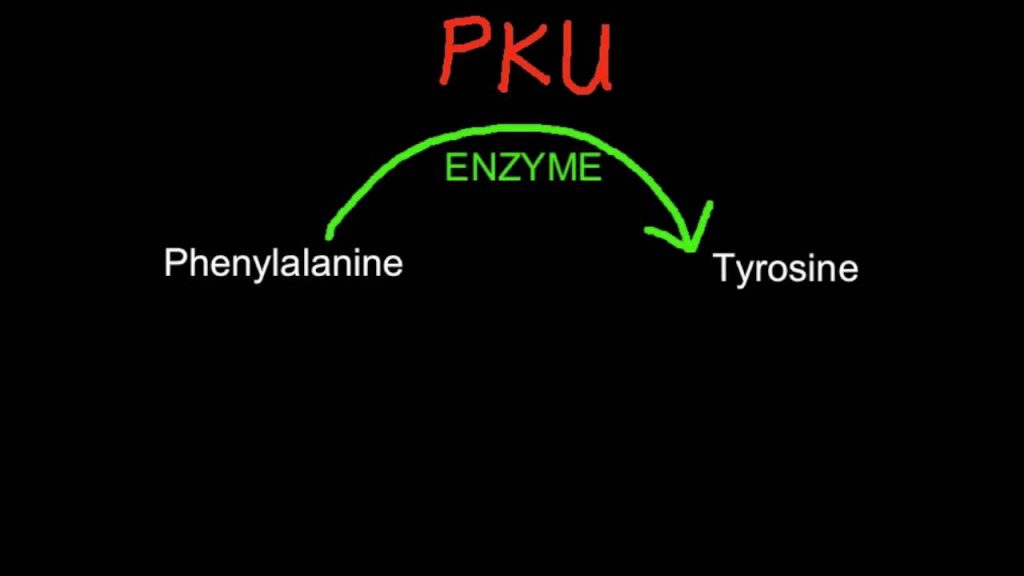
Η φαινυλκετονουρία, ICD-10 E70.0, είναι μία σχετικά σπάνια κληρονομική ασθένεια, που προκαλείται, όταν ο οργανισμός δε μπορεί να διασπάσει τη φαινυλαλανίνη. Η φαινυλαλανίνη είναι ένα από τα δομικά συστατικά των πρωτεϊνών στο σώμα μας. Όταν μπει στο σώμα, μέσω μίας σειράς αντιδράσεων, μετατρέπεται σε τυροσίνη, μία ουσία σημαντική για τη φυσιολογική λειτουργία του εγκεφάλου. Τα άτομα, λοιπόν, που πάσχουν από φαινυλκετονουρία αδυνατούν να πραγματοποιήσουν αυτή τη μετατροπή της φαινυλαλανίνης σε τυροσίνη, καθώς, το υπεύθυνο ένζυμο παρουσιάζει μειωμένη λειτουργία ή απουσιάζει εντελώς. Έτσι, η φαινυλαλανίνη σταδιακά συσσωρεύεται στον οργανισμό τους, με αποτέλεσμα την εμφάνιση ορισμένων αρνητικών συμπτωμάτων.
Η φαινυλκετονουρία είναι μία αυτοσωματική υπολειπόμενη γενετική μεταβολική διαταραχή, που χαρακτηρίζεται από ομόζυγες ή ετερόζυγες μεταλλάξεις στο γονίδιο για το ηπατικό ένζυμο υδροξυλάση φαινυλαλανίνης, που την καθιστά μη λειτουργική.
Το γονίδιο βρίσκεται στο χρωμόσωμα 12 στις ζώνες 12q22 – q24. Περισσότερες από 400 νοσογόνες μεταλλάξεις έχουν βρεθεί στο γονίδιο ΡΑΗ. Ανεπάρκεια ΡΑΗ προκαλεί ένα φάσμα διαταραχών από την κλασική φαινυλκετονουρία (PKU) και την υπερφαινυλαλανιναιμία (μία λιγότερο σοβαρή συσσώρευση φαινυλαλανίνη).
Όταν η δραστικότητα της υδροξυλάσης φαινυλαλανίνης είναι μειωμένη, η φαινυλαλανίνη συσσωρεύεται και μετατρέπεται σε φαινυλοπυροσταφυλικό (φαινυλκετόνη), η οποία μπορεί να ανιχνευθεί στα ούρα.
Με αυτοσωματική υπολειπόμενη διαταραχή και τα δύο αλληλόμορφα απαιτούνται, για να εμφανίσει ένα άτομα συμπτώματα της νόσου. Οι φορείς ενός αλληλόμορφου δεν εμφανίζουν συμπτώματα της νόσου, αλλά φαίνεται να προστατεύονται σε κάποιο βαθμό, έναντι της τοξίνης του μύκητα ωχρατοξίνη Α.
Υπερφαινυλαλανιναιμία από ανεπάρκεια της τετραϋδροβιοπτερίνης
Μία σπανιότερη μορφή υπερφαινυλαλανιναιμίας συμβαίνει, όταν το ένζυμο υδροξυλάση της φαινυλαλανίνης είναι φυσιολογικό, αλλά υπάρχει ένα ελάττωμα στη βιοσύνθεση ή την ανακύκλωση του συμπαράγοντα τετραϋδροβιοπτερίνη (ΒΗ4). Η ΒΗ4, που ονομάζεται βιοπτερίνη, είναι απαραίτητη για τη σωστή δραστικότητα του ενζύμου ΡΑΗ/Phenylalanine Hydroxylase/Υδροξυλάση της φαινυλανανίνης και αυτό το συνένζυμο μπορεί να συμπληρωθεί ως θεραπεία.
Η δραστικότητα της αναγωγάσης της διϋδροβιοπτερίνης απαιτείται, για την ανασύσταση της διϋδροβιοπτερίνης σε τετραϋδροβιοπτερίνη, που είναι ένας σημαντικός συμπαράγοντας σε πολλές αντιδράσεις στο μεταβολισμό των αμινοξέων. Εκείνοι οι ασθενείς, με αυτήν την ανεπάρκεια, μπορούν να παράγουν επαρκή επίπεδα του ενζύμου της φαινυλαλανίνης (PAH), αλλά δεδομένου, ότι η τετραϋδροβιοπτερίνη αποτελεί συμπαράγοντα, η ανεπάρκεια αναγωγάσης της διϋδροβιοπτερίνης η PAH, που παράγεται δε μπορεί να χρησιμοποιηθεί, για να παράγει φαινυλαλανίνη και τυροσίνη. Η τετραϋδροβιοπτερίνη είναι, επίσης, ένας συμπαράγοντας στην παραγωγή του L-DOPA από τυροσίνη και 5-υδροξυ-L-τρυπτοφάνης από την τρυπτοφάνη.
Η Τετραϋδροβιοπτερίνη απαιτείται για τη μετατροπή της φαινυλαλανίνης σε τυροσίνη, αλλά απαιτείται, επίσης, για τη μετατροπή της τυροσίνης σε L-DOPA, μέσω του ενζύμου υδροξυλάσης τυροσίνης. Η L-DOPA, στη συνέχεια, μετατρέπεται σε ντοπαμίνη. Χαμηλά επίπεδα της ντοπαμίνης οδηγούν σε υψηλά επίπεδα της προλακτίνης. Αντίθετα, στην κλασική φαινυλκετονουρία (χωρίς συμμετοχή διϋδροβιοπτερίνης), τα επίπεδα της προλακτίνης θα είναι σχετικά φυσιολογικά.
Ανεπάρκεια τετραϋδροβιοπτερίνης μπορεί να προκληθεί από ελαττώματα, σε τέσσερα διαφορετικά γονίδια (HPABH4A, HPABH4B, HPABH4C και HPABH4D).
Μεταβολικές οδοί φαινυλαλανίνης
Το ένζυμο υδροξυλάση φαινυλαλανίνης μετατρέπει, φυσιολογικά, το αμινοξύ φαινυλαλανίνη, στο αμινοξύ τυροσίνη. Εάν αυτή η αντίδραση δε λαμβάνει χώρα, η φαινυλαλανίνη και η τυροσίνη είναι ελλιπείς. Η υπερβολική φαινυλαλανίνη μπορεί να μεταβολιστεί σε φαινυλοκετόνες, μέσω μιας δευτερεύουσας οδού, μίας οδού με τρανσαμινάση γλουταμικού. Οι μεταβολίτες περιλαμβάνουν το φαινυλοξικό, φαινυλοπυροσταφυλικό και τη φαιναιθυλαμίνη. Αυξημένα επίπεδα της φαινυλαλανίνης στο αίμα και ανίχνευση των φαινυλοκετονών στα ούρα είναι διαγνωστικά, ωστόσο οι περισσότεροι ασθενείς διαγιγνώσκονται, μέσω της εξέτασης στη γέννηση.
Συμπτώματα φαινυλκετονουρίας
Τα συμπτώματα της νόσου περιλαμβάνουν βαριά νοητική καθυστέρηση, σπασμούς, διαταραχές συμπεριφοράς με στοιχεία αυτισμού, ποικίλες νευρολογικές διαταραχές, χαρακτηριστικό δερματικό έκζεμα και αποχρωματισμό του δέρματος, των μαλλιών και των ματιών.
Η νόσος εμφανίζεται, κλινικά, με επιληπτικές κρίσεις, υπερβολικά ξανθά μαλλιά και δέρμα και “οσμή μούχλας” από τον ιδρώτα του μωρού και τα ούρα (λόγω φαινυλοξικού, που είναι μία από τις κετόνες που παράγονται).
Διάγνωση φαινυλκετονουρίας
Η φαινυλκετονουρία μπορεί να ανιχνευθεί με τεστ αίματος, κατά τη γέννηση του νεογνού, το οποίο είναι τεστ ρουτίνας (συνήθως, πραγματοποιείται 2-7 ημέρες μετά τη γέννηση, χρησιμοποιώντας δείγματα, που λαμβάνονται από τσίμπημα του νεογνού στη φτέρνα).
Θεραπεία φαινυλκετονουρίας
Η πιο συχνή μορφή θεραπείας, για τα άτομα, που πάσχουν από φαινυλκετονουρία είναι η σωστή διατροφή.
Τα άτομα, αυτά, δεν πρέπει να παραλαμβάνουν με την τροφή τους μεγάλες ποσότητες φαινυλαλανίνης.
Οι τυπικές δίαιτες των ατόμων, που πάσχουν από φαινυλκετονουρία πρέπει να περιλαμβάνουν πολύ μικρές ποσότητες φαινυλαλανίνης και υψηλές ποσότητες των άλλων αμινοξέων και κυρίως, της τυροσίνης. Οι ποσότητες της επιτρεπόμενης φαινυλαλανίνης σε αυτά τα άτομα μπορούν να αυξάνονται σταδιακά, καθώς το παιδί μεγαλώνει.
Επομένως, στη δίαιτα αυτών των ατόμων επιτρέπονται:
Θρεπτικά συστατικά, όπως υδατάνθρακες, λίπη, βιταμίνες και ανόργανα συστατικά.
Τρόφιμα, με μέτριες ποσότητες πρωτεϊνών, όπως οι πατάτες, καθώς και τρόφιμα με χαμηλές ποσότητες πρωτεϊνών, όπως τα φρούτα, τα λαχανικά ή κατηγορίες ψωμιών με χαμηλή ποσότητα πρωτεϊνών.
Τα βέλτιστα επίπεδα φαινυλαλανίνης πρέπει να διατηρούνται μεταξύ 120μmol/L και 360μmol/L, κατά τη διάρκεια, τουλάχιστον, των δέκα πρώτων ετών. Όταν η φαινυλαλανίνη δε μπορεί να μεταβολιστεί από το σώμα, ασυνήθιστα υψηλά επίπεδα συσσωρεύονται στο αίμα και είναι τοξικά για τον εγκέφαλο. Όταν αφεθεί χωρίς θεραπεία, οι επιπλοκές περιλαμβάνουν σοβαρή νοητική αναπηρία, διαταραχές της λειτουργίας του εγκεφάλου, μικροκεφαλία, διαταραχές της διάθεσης, ακανόνιστη λειτουργία της κίνησης και συμπεριφορικά προβλήματα, όπως διαταραχή ελλειμματικής προσοχής.
Όλοι οι ασθενείς με PKU πρέπει να συμμορφώνονται με μία ειδική δίαιτα χαμηλή σε Phe, για τη βέλτιστη ανάπτυξη του εγκεφάλου.
«Διατροφή για μία ζωή» συνιστάται από τους περισσότερους ειδικούς.
Η διατροφή απαιτεί, σοβαρά, τον περιορισμό ή την εξάλειψη των τροφίμων με υψηλή περιεκτικότητα σε Phe, όπως το κρέας, το κοτόπουλο, τα ψάρια, τα αυγά, τα καρύδια, το τυρί, τα όσπρια, το γάλα και τα άλλα γαλακτοκομικά προϊόντα. Αμυλούχες τροφές, όπως οι πατάτες, το ψωμί, τα ζυμαρικά και το καλαμπόκι, θα πρέπει να παρακολουθούνται. Τα βρέφη μπορούν να συνεχίζουν να θηλάζουν, για να παρέχουν όλα τα οφέλη του μητρικού γάλακτος, αλλά θα πρέπει, επίσης, να παρακολουθούνται, για τα θρεπτικά συστατικά, που τους λείπουν και θα απαιτηθούν. Η γλυκαντική ουσία ασπαρτάμη, που υπάρχει σε πολλά τρόφιμα διατροφής και αναψυκτικών, πρέπει επίσης, να αποφεύγεται, καθώς η ασπαρτάμη περιέχει φαινυλαλανίνη.
Καθώς το παιδί μεγαλώνει, μπορεί να παίρνει ειδικά συμπληρώματα διατροφής και ειδικά σχεδιασμένα τρόφιμα. Επιπλέον, η τυροσίνη, η οποία κανονικά προέρχεται από φαινυλαλανίνη, πρέπει να συμπληρωθεί.
Η από του στόματος χορήγηση της τετραϋδροβιοπτερίνης (ή ΒΗ4), (ένας συμπαράγοντας, για την οξείδωση της φαινυλαλανίνης) μπορεί να μειώσει τα επίπεδα στο αίμα αυτού του αμινοξέος, σε ορισμένους ασθενείς. Έτσι, οι ασθενείς με φαινυλκετονουρία μπορούν να είναι σε θέση να αυξήσουν την ποσότητα των φυσικών πρωτεϊνών, με το να φάνε.
Διαιτητικά συμπληρώματα με μεγάλα ουδέτερα αμινοξέα (LNAAs/Large Neutral Amino Acids), με ή χωρίς την παραδοσιακή διατροφή φαινυλκετονουρίας είναι μία άλλη στρατηγική θεραπείας. Τα LNAAs (π.χ. Tyrosine, tryptophane, threonine, methionine, valine, isoleucine, leucine and histidine/Τυροσίνη, τρυπτοφάνη, θρεονίνη, μεθειονίνη, βαλίνη, ισολευκίνη, λευκίνη και ιστιδίνη) ανταγωνίζονται τη φαινυλαλανίνη, για συγκεκριμένες πρωτεΐνες φορείς μεταφοράς κατά μήκος του εντερικού βλεννογόνου στο αίμα και δια μέσου του αιματοεγκεφαλικού φραγμού στον εγκέφαλο. Μελέτες έδειξαν, ότι οι ασθενείς με φαινυλκετουρία, που παίρνουν, καθημερινά, συμπληρώματα LNAAs, έχουν μειωμένα επίπεδα φαινυλαλανίνης στο πλάσμα και μειωμένες εγκεφαλικές συγκεντρώσεις φαινυλαλανίνης, που μετρήθηκαν με φασματοσκοπία μαγνητικού συντονισμού.
Μία άλλη ενδιαφέρουσα στρατηγική θεραπείας, για ασθενείς με φαινυλκετονουρία, είναι το γλυκομακροπεπτίδιο καζεΐνη (cGMP/Guanosine 3′,5′-cyclic monophosphate/3′,5′-κυκλική μονοφωσφορική γουανοσίνη), που είναι ένα πεπτίδιο φυσικό χωρίς φαινυλαλανίνη. Το CGMP μπορεί να υποκαταστήσει το κύριο μέρος των ελεύθερων αμινοξέων στη διατροφή για φαινυλκετονουρία και παρέχει πολλές ευεργετικές θρεπτικές επιδράσεις, σε σύγκριση με τα ελεύθερα αμινοξέα. Το γεγονός, ότι το cGMP είναι ένα πεπτίδιο εξασφαλίζει, ότι ο ρυθμός απορρόφησης των αμινοξέων είναι παρατεταμένος, σε σύγκριση με τα ελεύθερα αμινοξέα και ως εκ τούτου, οδηγεί σε βελτιωμένη συγκράτηση πρωτεϊνών και αυξημένη αίσθηση κορεσμού, σε σύγκριση με τα ελεύθερα αμινοξέα. Ένα άλλο σημαντικό όφελος του cGMP είναι, ότι η γεύση βελτιώνεται σημαντικά, όταν το CGMP αντικαθιστά μέρος των ελεύθερων αμινοξέων και αυτό μπορεί να βοηθήσει στη βελτιωμένη συμμόρφωση με τη δίαιτα για τη φαινυλκετονουρία. Επιπλέον, το CGMP περιέχει ένα υψηλό ποσό LNAAs, που αποτελεί περίπου το 41g ανά 100g πρωτεΐνης και συνεπώς, θα συμβάλει στη διατήρηση των επιπέδων φαινυλαλανίνης πλάσματος στα επίπεδα στόχου.
Άλλες θεραπείες βρίσκονται υπό έρευνα, συμπεριλαμβανομένης της γονιδιακής θεραπείας και της θεραπείας υποκατάστασης του ενζύμου με αμμωνιακή λυάση της φαινυλαλανίνης (PAL/phenylalanine ammonia lyase).
Στο παρελθόν, οι συστάσεις για τη φαινυλκετονουρία ήταν η δίαιτα μέχρι 18 ετών. Σήμερα, οι περισσότεροι γιατροί συνιστούν στους ασθενείς με φαινυλκετονουρία δίαιτα σε όλη τους τη ζωή.
Όταν έχει η μητέρα φαινυλκετονουρία
Για τις γυναίκες, που πάσχουν από φαινυλκετονουρία, είναι απαραίτητη για την υγεία των παιδιών τους να διατηρούν χαμηλά επίπεδα φαινυλαλανίνης πριν και κατά τη διάρκεια της εγκυμοσύνης. Αν και το αναπτυσσόμενο έμβρυο μπορεί να είναι μόνο φορέας του γονιδίου φαινυλκετονουρίας, το ενδομήτριο περιβάλλον μπορεί να έχει πολύ υψηλά επίπεδα φαινυλαλανίνης, τα οποία μπορεί να διαπεράσουν τον πλακούντα. Το παιδί μπορεί να αναπτύξει συγγενή καρδιοπάθεια, καθυστέρηση της ανάπτυξης, μικροκεφαλία και πνευματική αναπηρία, ως αποτέλεσμα.
Στις περισσότερες χώρες, οι γυναίκες με φαινυλκετονουρία, που επιθυμούν να αποκτήσουν παιδιά καλούνται να μειώσουν τα επίπεδα φαινυλαλανίνης στο αίμα τους (συνήθως, μεταξύ 2mg/dL και 6mg/dL), πριν μείνουν έγκυες και προσεκτικά, να ελέγχουν τα επίπεδά τους, σε όλη την εγκυμοσύνη. Αυτό επιτυγχάνεται με τη διενέργεια τακτικών εξετάσεων αίματος και ακολουθώντας, αυστηρά, μία δίαιτα. Η ημερήσια πρόσληψη φαινυλαλανίνης της μητέρας μπορεί να διπλασιαστεί ή ακόμα και να τριπλασιαστεί, μέχρι το τέλος της εγκυμοσύνης, όταν αναπτύσσεται το ήπαρ του εμβρύου και παράγεται φαινυλαλανίνη. Όταν τα χαμηλά επίπεδα φαινυλαλανίνης διατηρούνται, κατά τη διάρκεια της εγκυμοσύνης, δεν υπάρχουν αυξημένα επίπεδα κινδύνου γενετικών ανωμαλιών.
Τα μωρά με φαινυλκετονουρία μπορούν να πίνουν μητρικό γάλα. Η αποκλειστική διατροφή με το μητρικό γάλα, για τα μωρά με φαινυλκετονουρία, μπορεί να τροποποιήσει τα αποτελέσματα της ανεπάρκειας, αν και κατά τη διάρκεια του θηλασμού, η μητέρα πρέπει να διατηρεί μία αυστηρή δίαιτα, για να κρατήσει τα επίπεδα της φαινυλαλανίνης χαμηλά.
Συχνότητα φαινυλκετονουρίας στις διάφορες χώρες:
Κίνα 1 σε 18.000
Φινλανδία <1 σε 100.000
Ιρλανδία 1 σε 4.500
Ιαπωνία 1 σε 120.000
Κορέα 1 σε 41.000
Νορβηγία 1 σε 13.000
Τουρκία 1 σε 2.600
Ινδία 1 σε 18.300
Ηνωμένες Πολιτείες 1 σε 15.000
Βιβλιογραφία
pmc.ncbi.nlm.nih.gov
Clin Biochem Rev. 2008 Feb;29(1):31–41.
Phenylketonuria: An Inborn Error of Phenylalanine Metabolism
Robin A Williams, Cyril DS Mamotte, John R Burnett
PMCID: PMC2423317 PMID: 18566668
Τα καλύτερα προϊόντα για τη φαινυλκετονουρία
Πατήστε, εδώ, για να παραγγείλετε τα καλύτερα προϊόντα για τη φαινυλκετονουρία

Διαβάστε, επίσης,
Ποιοι πρέπει να παίρνουν σπιρουλίνα
Είναι όλα τα πρόσθετα τροφίμων ανθυγιεινά;
Τι είναι το εμβρυϊκό υπερηχογράφημα


{kind=link}